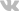
Механизмы инсулинорезистентности
 7909
7909  0
0  сахарный диабет
сахарный диабет 27.01.2014
27.01.2014 Консенсус конференции по инсулинорезистентности (1997) заключает: “Гиперинсулинемия является фактором, предопределяющим последующее развитие сахарного диабета. Понимание биологических аспектов инсулинорезистентности имеет большое значение для идентификации причинных генов и продуктов их экспрессии, а также для разработки новых методов терапии и оптимизации имеющихся методов лечения.
Основным аспектом инсулинорезистентности, интенсивно изучавшимся у людей, на животных моделях и клеточных культурах, является инсулинозависимое потребление и утилизация глюкозы. У больных с инсулинорезистентностью, это нарушение проявляется снижением инсулинозависимого накопления глюкозы в виде гликогена в мышцах и печени. На уровне мышечной ткани, предположительным первичным механизмом является блокирование транспорта глюкозы на ступени фосфорилирования. Этот дефект состоит из первичного генетического компонента и вторичного компонента факторов внешней среды.”
Особую роль играет снижение чувствительности к инсулину в мышечной, жировой и печеночной ткани, а также в надпочечниках. В миоцитах нарушается поступление и утилизация глюкозы, в жировой ткани развивается резистентность к антилиполитическому действию инсулина. Поступая в печень, СЖК, с одной стороны, становятся субстратом для формирования атерогенных липопротеидов, с другой – препятствуют связыванию инсулина с гепатоцитом, потенцируя ИР.
ИР гепатоцитов ведет к снижению синтеза гликогена, активации гликогенолиза и глюконеогенеза. ИР мышечной ткани проявляется в снижении поступления глюкозы из крови в миоциты и ее утилизации в мышечных клетках. ИР жировой ткани проявляется в резистентности к антилиполитическому действию инсулина, приводящему к накоплению свободных жирных кислот и глицерина. Анализ результатов современных исследований предполагает, что откладывание жира происходит не только в жировых депо, но и в других тканях, например, в скелетных мышцах и может способствовать развитию инсулинорезистентности, а откладывание липидов в β - клетках поджелудочной железы может нарушать их функцию, в конечном счете вызывая их гибель (Buckingham R.E. et al., 1998).
При нечувствительности ткани печени к действию инсулина усиливаются синтез глюкозы в печени и ее секреция в кровоток, более того, запускается гликогенолиз, а его образование и депонирование тормозятся. ИР ткани печени характеризуется активацией процессов неоглюкогенеза из аминокислот, лактата, пирувата, глицерина.
Stumvoll M. (1999) показал различия в степени чувствительности инсулинчувствительных тканей: минимальную степень ИР и у здоровых и при сахарном диабете 2 типа проявляет мышечная ткань, промежуточную – печеночная, а максимальную – жировая. У здоровых для подавления липолиза в жировой ткани на 50% требуется не больше 10 мкЕД/мл, для 50% подавления продукции глюкозы печенью необходимо уже около 30 мкЕД/мл, а для увеличения на 50% захвата глюкозы мышечной тканью дозу инсулина необходимо увеличить до 100 мкЕД/мл. При сахарном диабете 2 типа эта пропорция сохраняется на более высоких дозах инсулина соответственно 30, 50 и 120 мкЕД/мл.
Многочисленные исследования указывают на то, что гиперинсулинемия и хроническая передозировка инсулина при диабете способствует ИР [Lopez S.et al., 1983, Figlewicz D.P.et al., 1993].
В 1936 году Ланг и Лакенс показали, что при длительной гипергликемии, вызванной внутрибрюшинным введением глюкозы, происходит повреждение В-клеток и наступает стойкий СД.
Известно, что при 48-часовой инфузии 50% раствора глюкозы здоровым крысам, поддерживавшей гликемию выше 20 ммоль/л вызывало резкое снижение секреции инсулина. Инфузия липидов здоровым добровольцам в течение 48 часов приводила к ИР, хронической гипергликемии, которая сопровождалась гиперинсулинемией.
Описано возникновение гиперинсулинемии и следующей за ней инсулинорезистентности при ожоговой травме [Микаелян Н.П.1988], длительном воздействии гипокинезии [Карынбаев Ш.С. и др.,1982, Смирнов К.В., 1990], при краш - синдроме [Микаелян Н.П., 1990], инфаркте миокарда [Оганов Р.Г. и др., 1980], постреанимационном периоде [Сочнева Е.Н., 1994], при длительном курении [Fachini F.S. et al., 1995], пищевом рационе с высоким содержанием жира [Liu Sha et al.,1995].
ИР лежит в основе нарушения толерантности к углеводам [Остапова В.В., 1994]. Panay N. et al. (1997) обнаружили у здоровых женщин зависимость индекса резистентности к инсулину от фазы менструального цикла, показано его снижение в фоликуллярную фазу коррелирующее со снижением уровня прогестерона.
При СД 1 типа временная ИР может наблюдаться при декомпенсации диабета, кетоацидозе, инфекционных заболеваниях, а также после перенесенной гипогликемии. Достаточно продолжительная (на протяжении нескольких лет) ИР наблюдается в основном у больных пубертатного возраста и выражается в повышении потребности в инсулине более 1 ед/кг массы, достигающей в отдельных случаях 1,5 и даже 2 ед/кг. Одна из основных причин повышения потребности в инсулине при этом — увеличение секреции контринсулярных гормонов в период полового созревания и, в первую очередь, СТГ. Это является отражением физиологической ИР периода полового созревания, выражающейся в повышении уровня ИРИ и С-пептида в крови здоровых подростков [Кураева Т.Д, 2003].
ИР позволяет организму адаптироваться к гибельному воздействию гиперинсулинемии.. Показано, что сочетание 10-20 МЕД/кг инсулина с гипокинетическим стрессом приводило к гибели значительного числа крыс, уменьшению потребления ими пищи и появлению инсулинорезистентности [Носкович П. и др, 1991]. В наших экспериментах выживаемость крыс на фоне гипокинетического стресса значительно сокращалась при введении экзогенного инсулина в дозе 6 МЕД/кг [Макишева Р.Т., 1997].
ИР – полигенная патология, в развитии которой могут иметь значение мутации генов субстрата инсулинового рецептора (IRS-1 и IRS-2), b3-адренорецепторов, разобщающего протеина (UCP-1), а также молекулярные дефекты белков сигнального пути инсулина (глюкозные транспортеры). Мутации гена рецептора инсулина, которые приводят к ингибированию тирозинкиназы, сочетаются с резко выраженной инсулинорезистентностью. Аутофосфорилирование трех остатков тирозина (Tyr1146, Tyr1150, Tyr1151) усиливает активность киназ в 10-20 раз. Мутация одного или всех трех остатков тирозина приводит к резкому снижению инсулинстимулирующей активности киназ и параллельному снижению активности инсулина.
Мутации инсулинового рецептора подразделяют [Балаболкин М.И. 2000] на V классов:
- мутации, приводящие к снижению скорости биосинтезарецептора;
- мутации, ухудшающие внутриклеточный транспорт и посттрансляционный процессинг;
- мутации, приводящие к дефектам связывания инсулина;
- мутации, сопровождающиеся снижением рецепторной активности тирозинкиназы;
- мутации, ускоряющие деградацию инсулинового рецептора.
К механизмам тканевой инсулинорезистентности также относятся:
- отрицательная кооперативность связывания рецептора с инсулином, сопровождающаяся снижением сродства рецепторов к гормону в 10 раз, увеличением скорости диссоциации комплекса рецептор-гормон, снижением размеров солюбилизированного рецептора,
- усиление гликолиза рецепторов в аппарате Гольджи,
- уменьшение числа рецепторов инсулина вследствие их интернализации,
- уменьшение продолжительности жизни рецептора,
- нарушения синтеза рецепторов,
- уменьшение сродства рецептора к инсулину,
- образование антител к рецепторам инсулина. У больных ИЗСД в сыворотке крови определяются органоспецифические аутоантитела к тироглобулину, пероксидазе щитовидной железы, париетальным клеткам желудка, внутреннему фактору Кастла, клеткам коры надпочечника, антилимфоцитотоксические к тубулину, активину, иммуноглобулинам, и неорганоспецифические аутоантитела: антиядерные, к гладкомышечным волокнам, фибробластам, ретикулярные и митохондриальные.

Свежие статьи
- Купить Freestyle Libre
- Можно ли заменить уколы неинъекционными способами введения инсулина и глюкометрии?
- Неонатальный сахарный диабет
- Липоатрофический сахарный диабет
- Что такое предиабет и почему он возникает?

Читайте также:



